 移動端
移動端




 直播推薦
直播推薦
 回放
回放
 回放
回放


摘 要:研究了恒電流電解陰極銅使大部分銅與雜質分離,采用ICP-OES標準曲線法、基體匹配法和 多元光譜擬合(MSF)模型,同時測定陰極銅中Zn、Pb、Sn、Sb、Pb、Ni、Fe、P、As、Bi,并對電解液 中殘留銅量進行補正的方法,方法的檢出限為0.0008~0.047mg/L,回收率為90%~105%,RSD小于7.8%。 該法準確、快速、簡便,應用于陰極銅的測定, 結果滿意。
關鍵詞:陰極銅、恒電流電解、珀金埃爾默ICP-OES、基體匹配、多元光譜擬合
陰極銅中銅含量的測定在國家標準GB/T5121—2008中有詳細的介紹。電解-電解液中殘 留銅量補正法,雖然分析過程冗長,影響分析速度,不易控制,但是具有干擾少、準確度高 等優點。本文采用恒電流電解,電解液中殘留銅采用ICP-OES補正,同時電解液用于陰極銅 中Zn、Pb、Sn、Sb、Ni、Fe、P測定, As、Bi采用ICP-OES基體匹配法測定,能滿足日常陰 極銅的分析。
1、實驗部分
1.1 方法提要 試樣經酸分解, 在硝、硫混合酸介質中, 在陰、陽電極之間加上適當電流進行電解。電 解終止后,將沉積在鉑陰極上的金屬銅洗凈,烘干后稱量,電解液中殘留銅量用ICP-OES補正, 同時電解后的液體用于ICP-OES測定Zn、Pb、Sn、Sb、Pb、Ni、Fe、P, 其中As、Bi采用ICP-OES 基體匹配法測定。
1.2 主要儀器與試劑 1.2.1 44B型雙聯電解儀,上海雷磁創益儀器儀表有限公司 1.2.2 Optima 7300DV電感耦合等離子發射光譜儀(ICP-OES),美國PerkinElmer公司 1.2.3 鉑陰極(360~400孔/cm 2,Ф0.2~0.25 mm)將鉑陰極置于硝酸(1+1)中煮沸4~5min, 取出,用水沖洗干凈,取出,以無水乙醇浸洗二次,取出,用電吹風吹干,于干燥器內冷卻 后備用。經電解后的電極則需預先置于硝酸(1+1)中, 使銅*溶解后, 取出, 按上述步 驟處理后備用。 1.2.4 螺旋狀鉑陽極,處理同上。 1.2.5 混合酸:硝酸+硫酸+水=7 +10 +25 1.2.6 硝酸(1+1)1.2.7 醋酸(1+4) 1.2.8 無水乙醇,優級純 1.2.9 Cu、Zn、Pb、Ni、Fe、P、As、Bi標準溶液:1000ug/mL,Sn、Sb:500ug/mL(直接從 北京納克分析儀器有限公司購買) 1.2.10 高純銅基體溶液:稱取高純銅(99.999%)5.0000g,加入硝酸(1+1)30mL,蓋上表 面皿,低溫至*溶解,煮沸驅除黃煙,冷卻,用水沖洗表面皿及杯壁,加水稀釋至100mL, 此溶液濃度為50g/L。 1.2.11 Zn、Pb、Ni、Fe、P、Sn、Sb混合標準溶液:將標準溶液(1.2.9)稀釋為50ug/mL, 酸度為5%HNO3。 1.2.12 As、Bi混合標準溶液:將標準溶液(1.2.9)稀釋為50ug/mL,酸度為5%HNO3。
1.3 試驗方法 若試樣氧化,樣品應先于醋酸(1+4)中煮沸4-5min除去氧化層,用純水洗至無酸味, 再用無水乙醇浸洗并干燥后使用。 1.3.1 陰極銅中Cu的測定 稱取已處理好的試樣5.0000(±0.0002g)于電解杯中,緩緩加入混合酸(1.2.5)50mL, 蓋上表面皿,低溫至*溶解,煮沸驅除黃煙,冷卻,用水沖洗表面皿及杯壁,加水稀釋至 200mL,將已稱得其質量的鉑陰極、鉑陽極、攪拌子置于電解杯中,調節至適當攪拌速度, 用4A電流電解2h,電解結束,中斷電流,將電極架提起,使電極移開液面,用洗瓶沖洗陰極、 陽極表面,洗液接于原電極杯中,移開電解杯,再用洗瓶仔細沖洗電極表面,取下陰極在無 水乙醇中浸洗片刻,取出,用電吹風迅速吹干,置于干燥器中冷卻至室溫,稱得其質量。 洗液與電極杯中液合并于500mL容量瓶中,用ICP-OES測定殘留的銅量。 1.3.2 陰極銅中Zn、Pb、Sn、Sb、Ni、Fe的測定 上述(1.3.1)電解后的液直接用ICP-OES測定Zn、Pb、Sn、Sb、Ni、Fe、P。 1.3.3 陰極銅中As、Bi的測定 稱取已處理好的試樣0.5000(±0.0002g)于400燒杯中,加入硝酸(1+1)20mL,蓋上 表面皿,低溫至*溶解,煮沸驅除黃煙,冷卻,用水沖洗表面皿及杯壁,加水稀釋至100mL, 用ICP-OES測定。
1.4 儀器及工作條件 工作條件為:功率1300W,等離子體氣體流量15L/min,輔助氣體流0.2L/min,霧化器氣 體流量0.8 L/min,觀測距離15 mm,泵流量1.5mL/min,讀數延遲時間20S,自動積分時間1~ 5S,重復次數2次。 1.5 標準曲線繪制 1.5.1 移取0mL、1mL、2mL、3mL銅標準溶液(1.2.9),于一組100mL容量瓶中,分別加入10mL 硝酸(1+1),用水稀釋至刻度,混勻。1.5.2 移取0mL、0.8mL、1.2mL、2mL混合標準溶液(1.2.11),于一組100mL容量瓶中,分 別加入10mL硝酸(1+1),用水稀釋至刻度,混勻。 1.5.3 移取0mL、0.4mL、0.8mL、1.2mL混合標準溶液(1.2.12),于一組100mL容量瓶中, 分別加入10mL高純銅基體溶液(1.2.10)和10mL硝酸(1+1),用水稀釋至刻度,混勻。
2、結果與討論 2.1 測定條件的選擇 2.1.1 溶樣酸的選擇、用量、電解電流及電解時間的選擇 參考高學峰、張立婷、郭瑩著的電解銅中銅的快速測定方法,本法選用硝硫混合酸 (7+10+25)50mL。經試驗, 本法選擇電解電流密度4A/dm2,電解2h,殘留銅量用ICP-OES標準 曲線法補正,Cu譜線為327.393 nm,殘留量高達0.3%,仍可獲得很好的準確度與精密度。 2.1.3 Zn、Pb、Sn、Sb、Ni、Fe分析條件選擇 電解除去大部分銅基體后,電解液中殘留銅對Zn、Pb、Sn、Sb、Ni、Fe測定的干擾已經 很小,進行譜線掃描, 選擇干擾少的線作為分析譜線,Zn為206.200nm、Pb為283.306nm、Sn 為283.998nm 、Sb為206.836nm、Ni為341.476 nm、Fe為259.939 nm,直接用ICP-OES標準曲 線法測定。 2.1.4 P分析條件的選擇 雖然電解可以除去大部分銅基體,但是電解液中殘留銅對P的測定仍存在很大干擾,本 文采用多元光譜擬合(MSF)模型:用掃描方式,分別收集空白、分析物、干擾元素溶液的譜 圖。分析物濃度為100倍于檢出限以上,干擾物濃度為分析過程中可能遇到的高濃度,在 MSF編輯窗口中,設定每個元素的作用[空白(b)、分析物(a)、干擾物(i)],建立MSF模型, 選用P譜線 214.914 nm,用ICP-OES測定。 2.1.5 As、Bi分析條件的選擇 鉍(Ⅲ)和銅(Ⅱ)離子的還原電位值很接近,用控制陰極電位法難以達到兩者的分離 和測定,且通過實驗發現采用直接測定電解液中As,結果明顯偏高,故直接測定電解液中As、 Bi作為陰極銅的分析不可行。本文采用單獨溶樣,高純銅(99.999%)做基體,選用As譜線 188.979nm,Bi譜線190.171nm,用ICP—OES基體匹配法測定。
2.2 方法的準確度、精密度與回收率試驗 2.2.1 用國家標準物質純銅GBW02141,按試驗方法測定9次, 進行準確度和精密度試驗,結 果見表1。
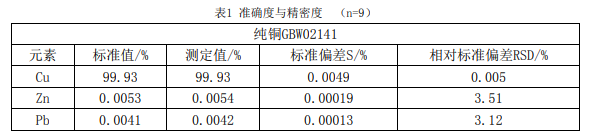
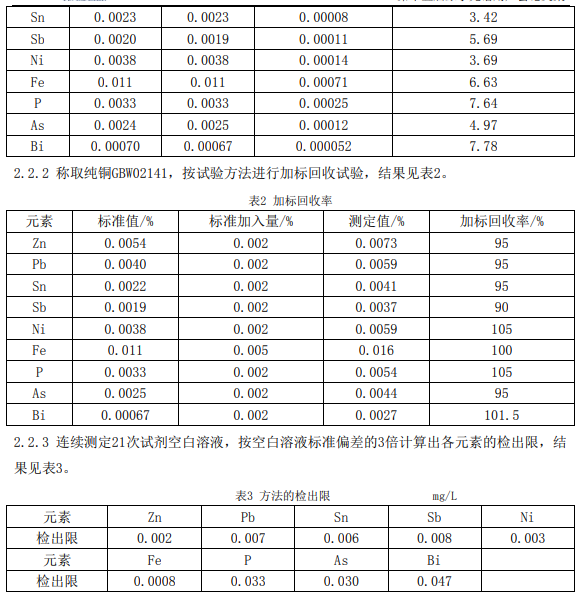
3、結論 本文研究討論了電解重量法與ICP-OES聯用測定陰極銅中銅及雜質元素的方法,從試驗 結果可以看出,測定結果準確可靠,采用本方法測定純銅GBW02141與其標準值相符,該方法 *可以滿足陰極銅日常分析。
參考文獻 [1]辛仁軒.等離子體發射光譜分析[M].北京:化學工業出版社, 2004.8 [2]符 斌,李華昌等.現代重金屬冶金分析[M].北京:化學工業出版社, 2006.10 [3]謝華林.基體分離ICP—AES法測定電解銅中雜質元素[J].冶金分析,2003,23(1):l2-13. [4]高學峰,張立婷,郭 瑩.電解銅中銅的快速測定[J].電鍍與環保,2003 [5]GB/T5121—2008,銅及銅合金化學分析方法[S].
免責聲明
- 凡本網注明“來源:儀器網”的所有作品,均為浙江興旺寶明通網絡有限公司-儀器網合法擁有版權或有權使用的作品,未經本網授權不得轉載、摘編或利用其它方式使用上述作品。已經本網授權使用作品的,應在授權范圍內使用,并注明“來源:儀器網”。違反上述聲明者,本網將追究其相關法律責任。
- 本網轉載并注明自其他來源(非儀器網)的作品,目的在于傳遞更多信息,并不代表本網贊同其觀點和對其真實性負責,不承擔此類作品侵權行為的直接責任及連帶責任。其他媒體、網站或個人從本網轉載時,必須保留本網注明的作品第一來源,并自負版權等法律責任。
- 如涉及作品內容、版權等問題,請在作品發表之日起一周內與本網聯系,否則視為放棄相關權利。


















 浙公網安備 33010602002722號
浙公網安備 33010602002722號
慕尼黑上海分析生化展(analytica China 2024)
展會城市:上海市展會時間:2024-11-18